Molecular Literacy Taskforce Terminology List
Glossary of Commonly Used Molecular Terms for Newborn Screening Follow-up Staff
Introduction: This glossary of molecular genetics terms is intended for use by Newborn Screening staff to help them achieve a working knowledge and richer understanding of genetics words, phrases and concepts that they are likely to come across in their day-to-day work. As the use of molecular testing is increasingly used by Newborn Screening Programs, staff are being called upon to communicate with laboratory staff, hospital staff, private physicians, specialists and even parents about genetic test results and test methods. The goal of this glossary is to facilitate a greater understanding of these terms and increase the confidence levels of staff when handling these communications.
If you have any additions you would like to suggest for this list please contact newsteps@aphl.org.
| A | B | C | D | E | F | G | H | I | J | K | L | M | N | O | P | Q | R | S | T | U | V | W | X | Y | Z |
Allele: /uh·leel/
Alleles are different versions of the same gene. Some alleles lead to obvious physical traits, symptoms, or diseases.
Amino Acid: /uh·MEE·no A·suhd/
The 20 amino acids are the building blocks of proteins in humans. Nine out of 20 cannot be made by the body and must be obtained through diet or supplementation, whereas the remaining 11 amino acids are made by the body. Based on the instructions provided by the mRNA (and the DNA) these amino acids form together to create protein. Amino acids are often referred to by their 3-letter abbreviation (e.g. Ala, Cys, etc.) or their 1-letter abbreviation (e.g. A, C, etc.).
| AMINO ACID | ABBREVIATION |
|---|---|
| Alanine (Ala) | A |
| Arginine (Arg) | R |
| Asparagine (Asn) | N |
| Aspartic acid (Asp) | D |
| Cysteine (Cys) | C |
| Glutamine (Gln) | Q |
| Glutamic acid (Glu) | E |
| Glycine (Gly) | G |
| Histidine (His) | H |
| Isoleucine (Ile) | I |
| Leucine (Leu) | L |
| Lysine (Lys) | K |
| Methionine (Met) | M |
| Phenylalanine (Phe) | F |
| Proline (Pro) | P |
| Serine (Ser) | S |
| Threonine (Thr) | T |
| Tryptophan (Trp) | W |
| Tyrosine (Tyr) | Y |
| Valine (Val) | V |
Cis and trans:
It is important to recognize that any genetic variants found may be in cis or in trans, as that may affect the phenotype of the disorder.
- Cis: /sis/
The variants are on the same chromosome. Two variants that are in “cis” may create a non-functional protein. As long as there are no variants on the opposing chromosome for that gene, the individual may have one functional protein. Depending on the disorder, the individual may show no symptoms of this disorder. - Trans: /tranz/
The variants are on opposing chromosomes, which may result in both genes forming non-functional proteins. An example of this may be the Q188R and the K285N variants of Galactosemia. If these two variants are found in cis, the individual would still have one functioning galactase gene, providing the enzyme ability to break down galactose; this baby would effectively be a carrier of Galactosemia and should not exhibit symptoms. However, if the variants are found in trans, the gene with Q188R results in a non-functional protein, while the gene with the K285N also results in a non-functional protein and the baby is expected to have symptoms of Classic Galactosemia. <
Chromosome: /KROW·muh·sowm/
A chromosome is a strand of DNA that is made up of hundreds to thousands of individual genes. Genes are usually located at specific locations within a chromosome.
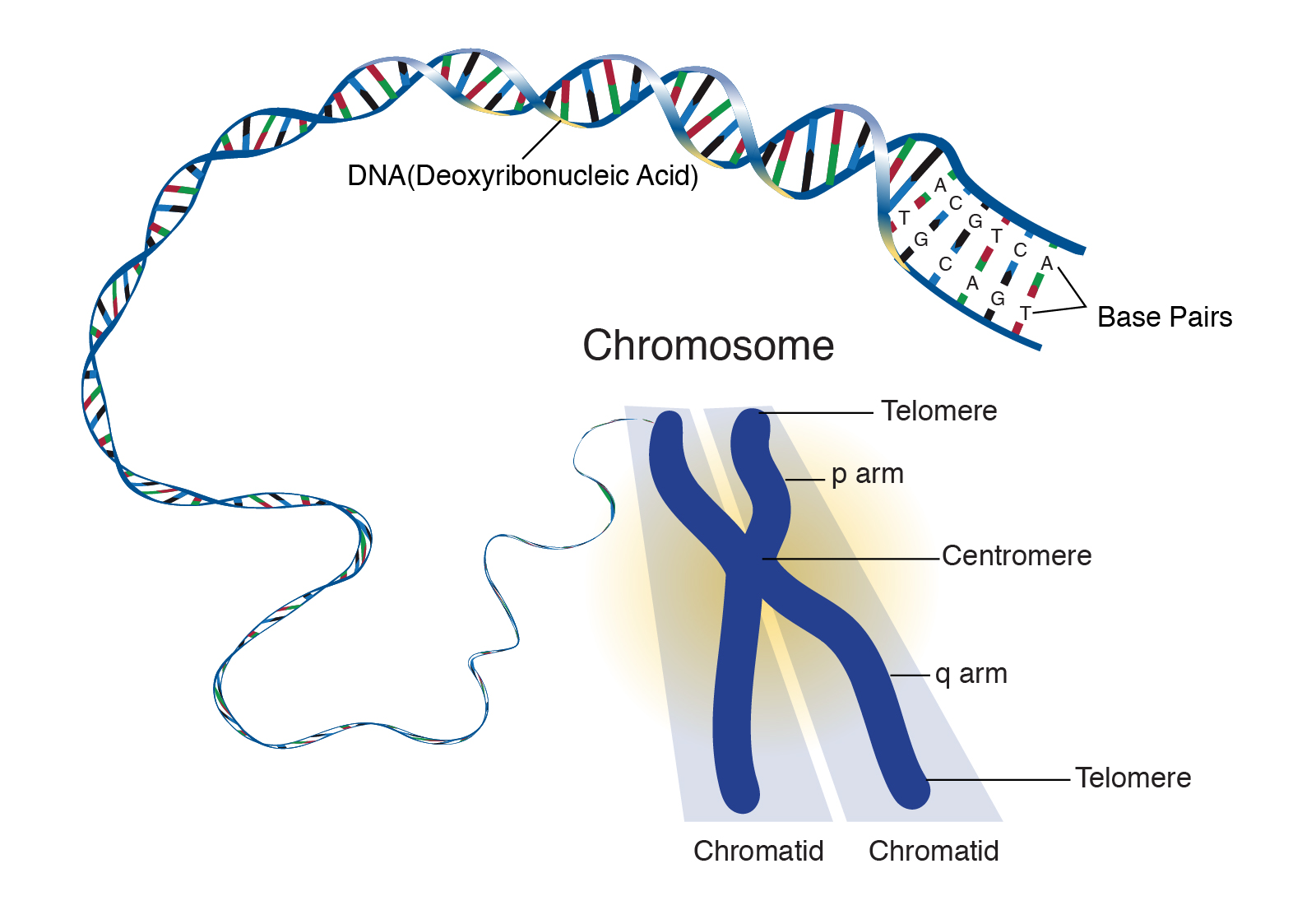 Source: National Human Genome Research Institute
Source: National Human Genome Research Institute
Humans typically have a total of 23 pairs of chromosomes (which includes 1 pair of sex, X and Y chromosomes). Typically, girls have two X chromosomes and boys have one X and one Y chromosome.
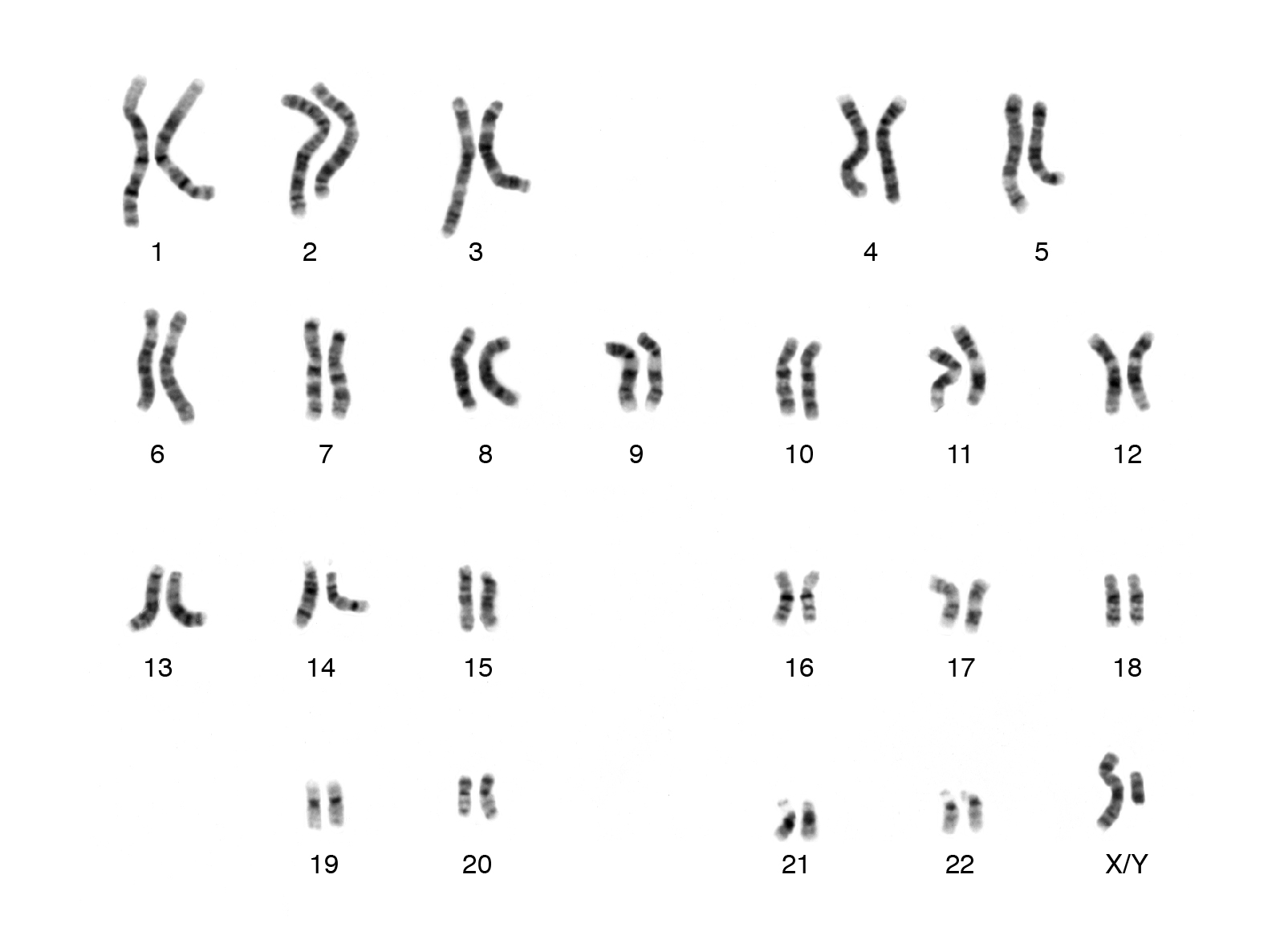 Source: National Human Genome Research Institute
Source: National Human Genome Research Institute
For each pair, we inherit one chromosome from our mother and one chromosome from our father.
Types of chromosome abnormalities:- Deletion: a part of the chromosome is missing.
- Duplication: a part of the chromosome is present multiple times.
- Translocation: a portion of one chromosome is transferred to another chromosome, or two chromosomes have switched pieces.
- Inversion: a portion of the chromosome has broken off, turned upside down, and reinserted.
- Insertion: a portion of one chromosome has been inserted into another chromosome.
- Rings: a portion of a chromosome has broken off and formed a circle or ring. This can happen with or without loss of genetic material.
- Isochromosome: Formed by the mirror image copy of a chromosome segment including the centromere (hyperlink here).
Missing, duplicated, or defective genes can result in loss of protein production or an abnormal protein. This can then result in physical symptoms, traits or diseases.
Common mutation panel:
A genetic test that includes a limited number of the most common disease-causing variants in a particular gene. The benefits of this approach are that it has a quick turnaround time, is cheaper, and the test result is relatively easy to interpret. The limitations of this approach are that it is not comprehensive, meaning that a baby may have other variants in that gene besides those tested for in the panel and these will not be detected. Common mutation panels are used frequently in CF screening as a second-tier test, but may be used with other disorders as well.
Deletion/duplication testing:
A genetic test that involves looking for large deletions and/or duplications of DNA within a gene. This is usually done as a supplemental test to full gene sequencing or other genetic testing and would be ordered by a specialist.
Deoxyribonucleic acid (DNA): /dee·OK·si·RAHY·boh·noo·KLEE·ik A·suhd/
DNA is a molecule that contains information needed to build and maintain our bodies. This information is found inside every cell and is passed down from parents to their children. It is considered to be one of the building blocks of our body. It is composed of 4 nucleotides (or “bases”) Adenine (A); Thymine (T); Cytosine (C); Guanine (G). These four bases form genes that may be hundred to thousands of bases long. For example, the CFTR gene for Cystic Fibrosis is 189,000 bases long, while the GALT gene for Galactosemia is 4,450 bases long. Changes or alterations in the DNA may cause changes in the protein or enzyme formation, which may lead to altered or loss of function of a critical protein.
Two strands of nucleotides (bases) are attached together based on “complementary bases”. An A and a T base complement and a C and a G base complement. These two strands form the DNA double helix.
Full gene sequencing:
A genetic test that involves reading through the full DNA sequence of a particular gene. The benefit of this approach is that it is more comprehensive and will detect the majority of potential variants in that gene. The limitations to this approach are that it takes longer (both the testing and interpretation), is more expensive, and there is a possibility of ambiguous test results (ie. variants of uncertain significance). An additional limitation of full gene sequencing is that it may not be able to detect large deletions or duplications of DNA in the gene, so still not fully comprehensive. Full gene sequencing may be done as a confirmatory diagnostic test for many of the disorders screened for by NBS Programs; it may also be used as a second or third-tier test for conditions such as Pompe disease.
- Sequencing is one of the ways that we can look at genes to find differences that cause genetic conditions. The genetic alphabet has only 4 letters: A, T, C, and G. Each gene is made up of a combination of these letters and may be hundreds to thousands of letters long. Each gene must have all of the letters in the correct order to work normally within the body. Differences in the spelling of a gene (which are often called variants) can be detected through genetic testing, which is usually done with a blood test. Sequencing tests can only look for spelling variations or single letter changes within a gene and are not able to detect other differences such as groups of letters that have been deleted or duplicated. Depending on what the type of sequencing, testing may look at one gene at a time or a group of genes.
- In comparison, biochemical screening or testing looks at the consequences of genetic variants or changes. Having an abnormal biochemical tests does not always mean that there is a genetic difference in the individual. Biochemical tests are often used as a first tier test in newborn screening, and genetic testing may be done as either a second tier or diagnostic follow-up.
Gene: /jeen/
Segments of DNA that provide our bodies with instructions on how to make proteins. The Human Genome Project has estimated that humans have between 20,000 and 25,000 genes. With the exception of the X and Y chromosomes, every person has two copies of each gene, one inherited from each parent. Most genes are the same in all people, but a small number of genes (less than 1 percent of the total) are slightly different between people.
- Coding genes are the instructions given by genes to form proteins. Proteins do most of the work in cells and are required for the structure and function of the body's tissues and organs. Noncoding genes are those sections of DNA that do not provide instructions to make proteins and/or are not expressed.
Genotype: /JEEN·uh·taip/
The genetic makeup of an individual.
- gene combination at one specific locus or any specified combination of loci.
- the alleles situated at one or more sites on homologous chromosomes; often used when referring to the combination of alleles located on homologous chromosomes that determines a specific characteristic or trait.
- Genotype vs. Phenotype: The difference between genotype and phenotype often comes up when discussing conditions on newborn screening panels. Sometimes the genotype is strongly associated with a specific phenotype. Therefore, knowing the genotype may help predict the expected course of a condition. An example would be in Cystic Fibrosis, genotype ΔF508/ΔF508, would be associated with pancreatic insufficiency. However, genotypes may not predict phenotypes in other conditions. An example would be X-ALD. Brothers with the same X-linked variant of the ABCD1 gene may not experience the same course of disease. One may have the early onset cerebral form while his brother has a later onset adrenomyeloneuropathy.
Hemizygous: /HEH·mee·ZY·gus/
Having only one copy of a gene (humans have two chromosomes, so it is more common to have two copies of each gene). The biggest example of this is that males have only one copy of each of the genes found on the sex chromosomes, as they inherit an X and a Y chromosome. An example of this is baby boys who screen positive for X-Linked Adrenoleukodystrophy and are found to be hemizygous for an ABCD1 pathogenic variant on their single X chromosome.
Heredity: /her·EH·duh·tee/
The transmission of characteristics from one generation to the next. Examples include:
- Autosomal Dominant: one chromosome with a gene that has a variant which causes a disease or trait to occur. The copy of the same gene on the other chromosome is normal. This variant is inherited from one parent.
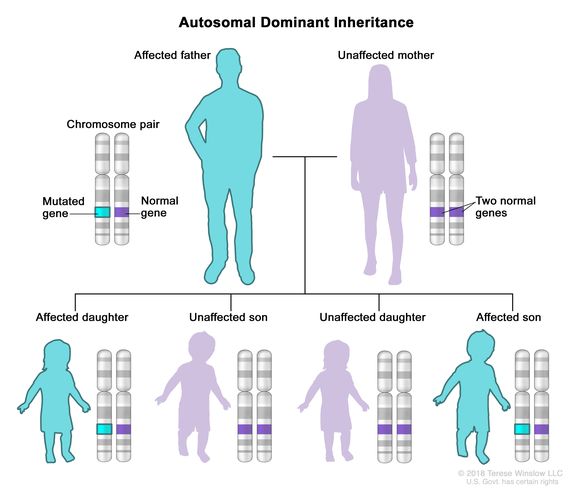 Source: National Cancer Institute
Source: National Cancer Institute
- Autosomal Recessive: the same gene on both chromosomes have a variant which causes a disease or trait to occur. One copy of the variant is inherited from each parent.
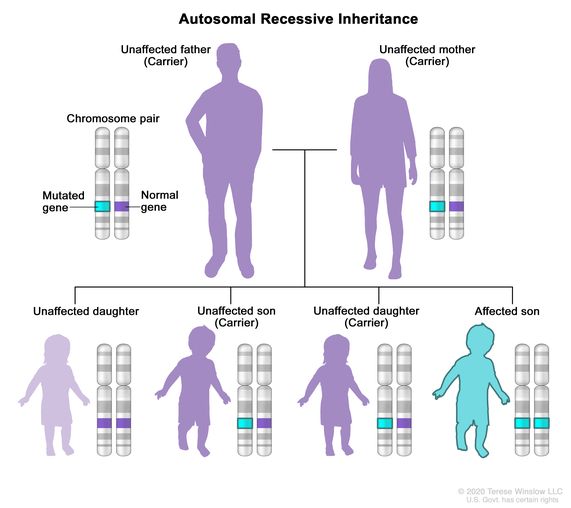 Source: National Cancer Institute
Source: National Cancer Institute
- De Novo: a variant in a gene that is present for the first time in one family member. The variant is new and was not inherited.
- X-linked: the gene variant is located on the X chromosome
Heterozygous: /HEH·tr·ow·ZAI·guhs/
When a person inherits two different alleles or variants of a gene. This may be in any combination of the American College of Medical Genetics and Genomics (ACMG) variant classifications. For example, in babies who screen positive for Galactosemia, they may have two pathogenic variants of Q188R/K285N; a pathogenic and a benign Q188R/N314D; or a pathogenic and a normal copy of Q188R/wildtype (normal).
Homozygous: /HO·mow·ZAI·guhs/
When a person inherits two copies of the same allele or variant of a gene. An example of this is the Q188R/Q188R genotype for Galactosemia. Q188R is the most common pathogenic variant and some individuals with Galactosemia have two copies of this variant. A second example is the ΔF508/ΔF508 allele for Cystic Fibrosis.
Locus: /LOW·kuhs/ (pl. loci /LOW·sai/)
The specific physical location of a gene on a
chromosome.
Mutation: /myoo·TEI·shn/
See Variant.
Nomenclature: /NOW·muhn·klei·chr/
Nomenclature – variants may have several names, depending on the nomenclature used. There is information “hidden” within each of the names for all of the nomenclature types. The CFTR variant is known by G551D, p.Gly551Asp, and c.1652G>A.
- G551D – common name
- G – indicates the amino acid in the “normal” or “wildtype” of the protein. G is the one letter abbreviation for Glycine.
- 551 – indicates the location of the altered amino acid in the protein sequence
- D – indicates the type of amino acid in the altered protein sequence. The D is the one letter abbreviation for Aspartic Acid.
- What this means, is that at location 551 a glycine was changed to an aspartic acid.
- p.Gly551Asp
- p. – indicates we are looking at the protein nomenclature
- Gly - indicates the amino acid in the “normal” or “wildtype” of the protein. The “Gly” is the three letter abbreviation for Glycine.
- 551 – indicates the location of the altered amino acid in the protein sequence.
- Asp – indicates the type of amino acid in the altered protein sequence. “Asp” is the three letter abbreviation for Aspartic Acid.
- What this means, is that at location 551 a glycine was changed to an aspartic acid.
- c.1652G>A
- c. – indicates that we are looking at the DNA nomenclature. The c references the coding DNA sequence. Note that a “g.” in this location would refer to the genomic DNA sequence.
- 1652 – indicates the location in the gene where we can find the variant
- G – indicates the DNA nucleotide in the “normal” or “wildtype” of the DNA. Note that the “G” indicated here is for the Guanosine nucleotide found in DNA, not the “G” amino acid glycine.
- > - the type of nucleotide change. The “>” indicates a substitution of one nucleotide for another.
- A – indicates the DNA nucleotide Adenosine that was found.
- What this means, is that at location 1652 a Guanosine was changed to an Adenosine nucleoside.
Phenotype: /FEE·now·taip/
Any observable or identifiable structural or functional characteristic of an organism. The expression of a specific trait, such as eye color or blood type, based on genetic and environmental influences.
- Genotype vs. Phenotype: The difference between genotype and phenotype often comes up when discussing conditions on newborn screening panels. Sometimes the genotype is strongly associated with a specific phenotype. Therefore, knowing the genotype may help predict the expected course of a condition. An example would be in Cystic Fibrosis, genotype ΔF508/ΔF508, would be associated with pancreatic insufficiency. However, genotypes may not predict phenotypes in other conditions. An example would be X-ALD. Brothers with the same X-linked variant of the ABCD1 gene may not experience the same course of disease. One may have the early onset cerebral form while his brother has a later onset adrenomyeloneuropathy.
Polymerase chain reaction (PCR): /puh·LI·mr·eis/
There are often too few copies of DNA within a sample for molecular biologist to be able to identify variants. PCR is a technique used to make many copies of a specific segment of DNA (also known as PCR amplification). The process is repeated many times for exponential numbers of DNA copies.
Protein: /PRO·teen/
Large molecules composed of amino acids, as determined by the DNA sequence. Proteins are required for the structure, function and regulation of the body’s cells, tissues and organs. Proteins have a variety of functions including:
- antibodies for immunological responses
- enzymes to create chemical reactions and/or form new molecules (e.g. GALC protein associated with Krabbe, GALT protein associated with Galactosemia)
- coordination between different cells (e.g. hormones, CFTR protein associated with cystic fibrosis, or SMN protein associated with SMA)
- provide structural support for cells and for movement of the body
- transport and/or storage of other small molecules (e.g. hemoglobin associated with sickle cell disease, GAA protein associated with Pompe)
Pseudodeficiency Allele: : /SOO·dow·duh·FI·shuhn·see uh·LEEL/
A variant or change in the DNA sequence of a gene that is known to change the gene’s expression but without causing disease. Babies may have one or more pseudodeficiency alleles only, or in combination with other gene variants. Pseudodeficiency alleles are very common in Pompe disease, as well as some other conditions. In Pompe disease, pseudodeficiency alleles may appear to lower enzyme activity but do not cause disease.
Pseudogene : /SOO·dow·jeen/
An imperfect copy of a functional gene. This may result in a non-functional or poorly functioning protein. An example of this is CAH which has multiple pseudogenes which may complicate molecular analysis and clinical diagnosis.
Ribonucleic acid (RNA): /RAHY·boh·noo·KLEE·ik A·suhd/
Exists in every cell in the body and it helps to translate DNA into protein.
Trans : See Cis and trans.
Variant: /VEH·ree·uhnt/
The word “mutation” has historically been used to describe changes in DNA sequence which are rare and disease causing. In an effort to be more consistent, genetic changes are now referred to as “variants”, and may include the American College of Medical Genetics and Genomics (ACMG) variant classifications of Pathogenic, Likely Pathogenic, Variant of Unknown/Uncertain Significance (VUS/VOUS), Likely Benign or Benign.
ACMG guidelines for DNA variants
In 2015, the American College of Medical Genetics (ACMG) published guidelines to assist in the classification in variants. This guideline has a list of recommended population and disease specific databases to begin the process of classifying variants in relation to their consequence and influence in causing disease. The 5 classifications are:
- Pathogenic – the variant directly contributes to the disease or disorder.
- Likely Pathogenic – it is highly likely that the variant contributes directly to the disease or disorder, however additional evidence is needed.
- Variant of Uncertain Significance (VUS) – an identified variant whose contribution to the disease or disorder is not known.
- Likely Benign – it is highly likely that the variant does not contribute to the disease or disorder, however additional evidence is needed.
- Benign – the variant does not contribute to disease.